Genetic Diversity and Evolutionary Dynamics of Influenza A and B Viruses

Influenza viruses are members of the Orthomyxoviridae family whose genomes consist of 8 segments of single-stranded negative-sense RNA. Influenza A virus (IAV) and influenza B virus (IBV) are responsible for seasonal epidemics of respiratory illness among humans. While IBV circulates only among humans, IAV circulates among many mammalian and avian hosts. Global surveillance of these viruses is necessary for maintaining a well-matched annual influenza vaccine, as well as for detecting the emergence of new influenza strains that may cause a human pandemic. Hemagglutinin (HA) and neuraminidase (NA) are the two predominant viral glycoproteins found in the viral envelope, and are the primary targets of the host immune system and are key factors in the entry and exit of influenza particles from host cells. HA and NA are also used to classify influenza A viruses into subtypes (e.g., H3N2, H5N1).
Our collaborative influenza projects will: 1) characterize the temporal and spatial evolutionary dynamics of the IAV and IBV circulating globally in humans and wild birds, 2) identify the IAV genotypes circulating in pigs at county fairs (swine-human interface) to determine if fairs produce novel reassortants or enable zoonosis/reverse zoonosis, 3) determine if specific internal gene segment constellations enhance viral fitness, 4) detect epistatic interactions between gene segments, and 5) assess the impact of inter-hemispheric migration of viruses/gene segments.
The accumulation of mutations at antigenic sites (antigenic drift) and the reassortment of segments with differing evolutionary histories (antigenic shift) allow influenza viruses to continually evade the host immune response. Annual human epidemics are currently caused by two IAV subtypes (A/H3N2, A/H1N1) and by two diverging IBV lineages (B/Yamagata, B/Victoria). IAVs circulating in avian and swine hosts are also important because of frequent interspecies transmission, which periodically causes human pandemics (e.g., pandemics of 1957, 1968, and 2009). Complete genome sequencing is required to identify fitness advantages conferred by genes and how antigenic drift in HA/NA drive mutations throughout the genome. Genomic sequence data will be generated from the diverse subtypes/strains (H1-H16, N1-N9) in avian reservoirs, which are a source of viruses with pandemic potential (e.g., H5N1). Swine are 'mixing vessels' for reassortment between avian and human viruses because they express sialic acid molecules that act as receptors for both avian and human viruses. Human, avian, and classical swine lineage viruses are co-circulating in North American swine populations, generating novel reassortants and leading to hundreds of zoonotic infections (e.g., H3N2v). The large number of co-circulating viruses in swine is the most genetically dynamic situation observed in North America over the last 90 years.
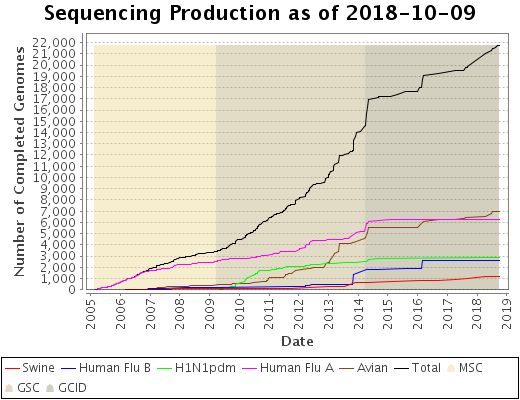
Status Graph
Funding
Research reported in this publication was supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number U19AI110819. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.